Signaling Pathways to Cell Motility
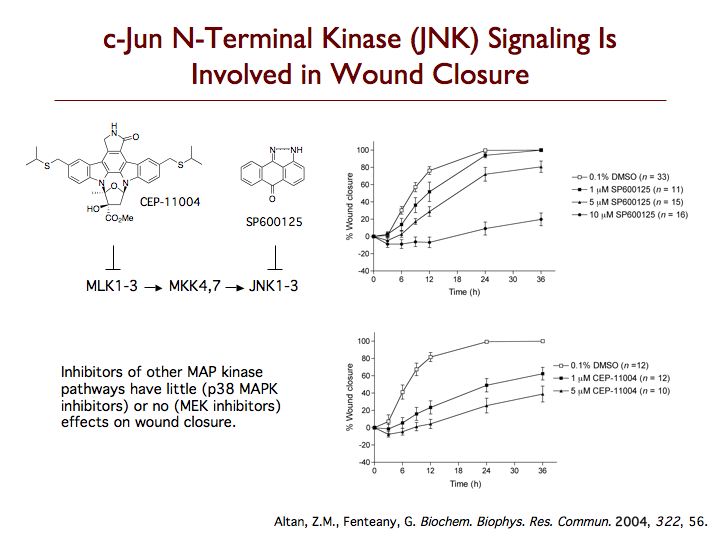
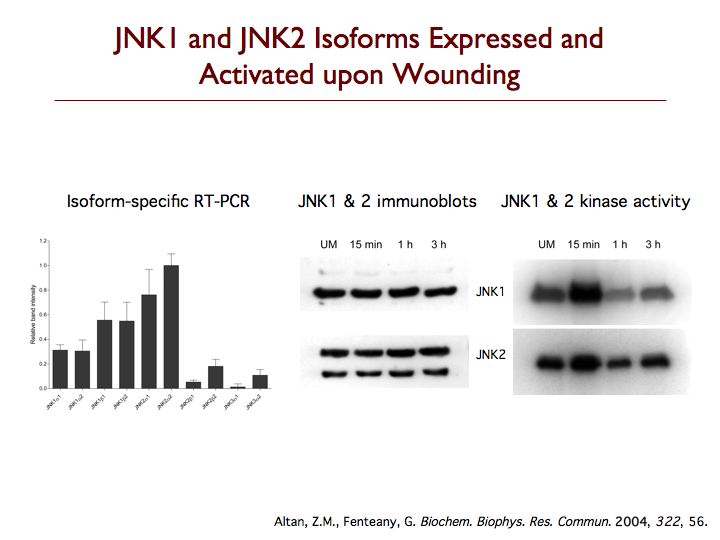
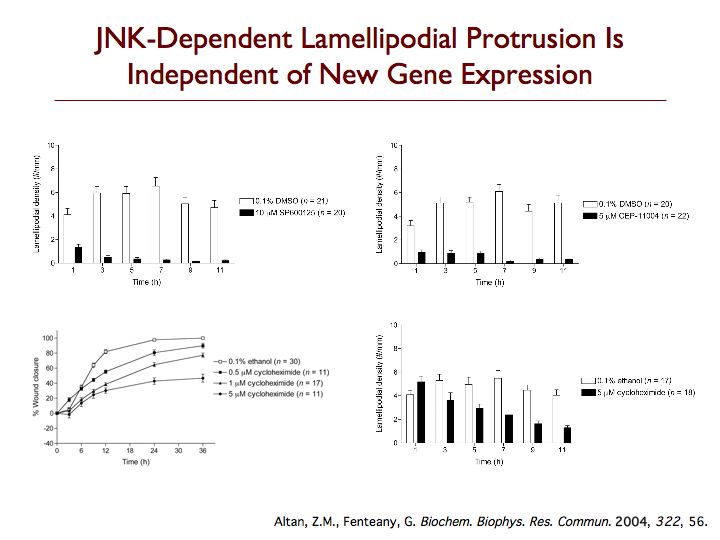
We are exploring the pathways leading to cell motility, particularly during Madin-Darby canine kidney (MDCK) epithelial cell sheet migration, looking at the signaling proteins that trigger the collective movement of cells that drives wound closure. One protein that is critical to this process is c-Jun N-terminal kinase (JNK), which is emerging as an important regulator of cell migration. Perturbing the JNK signaling pathway with three structurally and mechanistically distinct inhibitors that selectively target either JNKs themselves or the upstream mixed-lineage kinases, we found dramatic inhibition of membrane protrusion and cell sheet migration during wound closure in MDCK cell monolayers. Extension of lamellipodia is blocked from the earliest times after wounding in the presence of JNK pathway inhibitors, whereas assembly of non-protrusive actin bundles at the wound margin is unaffected. Inhibitors of the other mitogen-activated protein kinase (MAPK) pathways, the extracellular signal-regulated kinase and p38 MAPK pathways, only have comparatively weak or marginal inhibitory effects on wound closure. Multiple splice variants of both JNK1 and JNK2 are expressed in MDCK cells, and JNK1 and JNK2 are rapidly and transiently activated upon wounding. Phosphorylation of c-Jun does not appear relevant to MDCK wound closure, and membrane protrusion directly after wounding is not affected by inhibitors of RNA or protein synthesis. While most known substrates of JNK are transcription factors or proteins regulating apoptosis, our data indicate that JNK regulates protrusion and migration in a gene expression-independent manner and suggest an important cytoplasmic role for JNK in the control of cell motility.
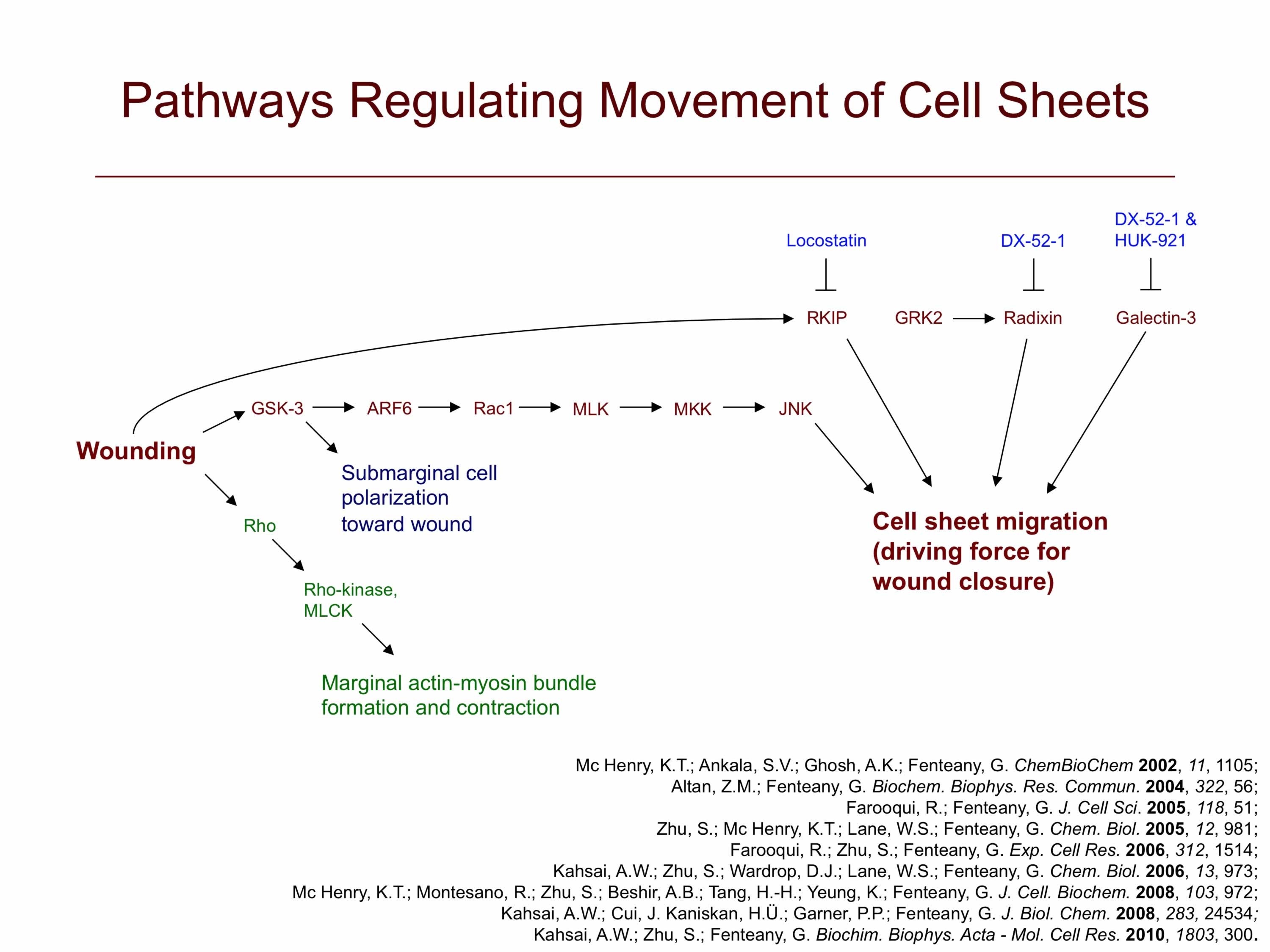
In addition, we have found that glycogen synthase kinase-3 (GSK-3) activity is required for membrane protrusion and crawling of cells at the wound edge and those behind it in wounded MDCK cell monolayers. RNA interference-based silencing of GSK-3α and GSK-3β expression also results in slowed cell sheet migration, with the effect being more pronounced with knockdown of GSK-3β. Both GSK-3α and GSK-3β are in activated states during the most active phase of cell migration. In addition to having a positive control or permissive, rather than negative, function in MDCK cell migration, GSK-3 appears to act upstream of the small GTPases ADP-ribosylation factor 6 (ARF6) and Rac1. Expression of constitutively active ARF6 restores a protrusive, migratory phenotype in cells treated with GSK-3 inhibitors. It does not, however, restore to normal levels the directional polarization of cells behind the wound edge toward the wound area, implying the existence of a separate ARF6-independent branch of the GSK-3 pathway that regulates proper wound-directed polarization of these cells. Finally, inhibition of GSK-3 also strongly reduces activation of Rac1 and cell scatter in response to hepatocyte growth factor/scatter factor, which triggers dispersal and migration of cells in monolayer culture as fibroblast-like individual cells, a mode of epithelial cell motility distinct from the collective migration of wound closure.
We are further exploring this pathway and working on the discovery of new small-molecule probes of cell motility to help dissect the signaling cascades that control cell sheet migration. For instance, using a cell migration inhibitor we discovered, locostatin, we implicated Raf kinase inhibitor protein in the control of cell sheet migration.
More on the mechanism of cell sheet migration.